AFToL and WASABI
WASABI – Web Accessible Sequence Analysis for Biological Inference:
The AFToL database interface.
F. Kauff, C. J. Cox, and F. Lutzoni
‘Fungal trees grow faster with computer help’ – Science, Vol 309, Issue 5733, 374 , 15 July 2005.
(click here for PDF format)
Introduction
The goal of the NSF-funded AFToL (Assembling the Fungal Tree Of Life) project is to enhance the understanding of the evolution of the Kingdom Fungi by sampling 1500+ species for eight gene loci across all major fungal clades, plus a subset of taxa for a suite of morphological and ultrastructural characters. Fungi play pivotal ecological roles: as saprotrophs, they are important in the cycling of nutrients; as pathogens and parasites, they attack virtually all groups of organisms, including bacteria, plants, other fungi, and animals; as mutualistic symbionts, fungi have enabled a diversity of other organisms to exploit novel habitats and resources. Fungi have been found in every ecosystem where they have been sought, including deserts, glacial ice, and deepsea thermal vent communities. For these reasons, a robust fungal phylogeny will greatly enhance our understanding of the history of life.
AFToL is a collaboration centered around four universities in the United States: Duke University (Francois Lutzoni and Rytas Vilgalys), Clark University (David Hibbett), Oregon State University (Joey Spatafora), and University of Minnesota (David McLaughlin). Participants throughout the world have donated vouchers, taxon samples, and gene sequences. The aim of the project is to reconstruct the fungal tree of life using all available data for eight loci [nuclear ribosomal DNA: LSU, SSU, ITS (including 5.8s, ITS1 and ITS2); RNA polymerase II: RPB1, RPB2; elongation factor 1-alpha; mitochondrial SSU rDNA, and mitochondrial ATP synthase protein subunit 6 ]. A further objective of this study is to summarize and integrate our current knowledge regarding fungal subcellular features within this new phylogenetic framework.
Communication Framework
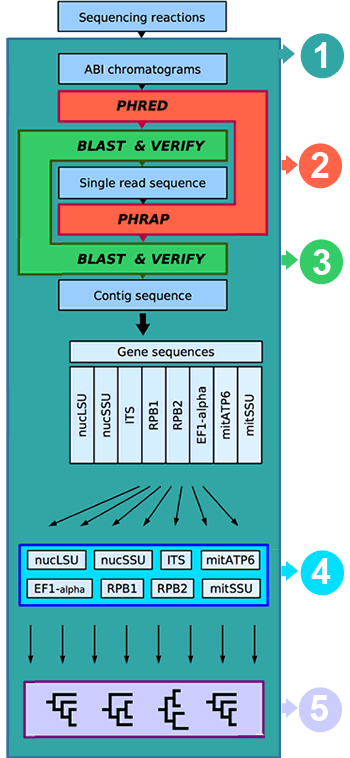
The name of the bioinformatic package we developed for AFToL is WASABI (Web Accessible Sequence Analysis for Biological Inference). WASABI provides an efficient communication platform to facilitate the collection and dissemination of molecular data to (and from) the laboratories and participants. All molecular data can be viewed, downloaded, verified, and corrected by the participants of AFToL. A central goal of the WASABI interface is to establish an automated analysis framework that includes basecalling of newly generated chromatograms, contig assembly, quality verification of sequences (including a local BLAST), sequence alignment, and congruence test. Gene sequences that pass all tests and are finally verified by their authors will undergo automated phylogenetic analysis on a regular schedule. Although all steps are initially carried out noninteractively, the users can verify and correct the results at any step and thus initiate the reanalysis of dependent data.
All custom made software applications are written in Python (www.python.org) and regularly use modules provided by BioPython (www.biopython.org).
(1) Web interface and SQL database
All data, including taxon and voucher information, gene sequences, and intermediary results, are stored in an SQL database. Access to the data is provided for registered users using a secure web interface build upon the Zope application server (www.zope.org). Participants of AFToL use the interface to enter specimen voucher information, to maintain sequencing primer information, and to access and verify the results of their sequencing reactions, automated analyses, and blast searches during all stages of the automated analyses.
The wwwinterface also provides functionality to include GenBank sequences for further analysis and to faciliate submission of finalized gene sequences to GenBank.
The database and web interface are closely linked to a set of software applications which analyze and verify the user generated sequence data. Each time sequences are entered, corrected, or deleted, subsequent analyses related to these changes are triggered to keep all information in the database consistent.
(2) Base-calling and contig assembly
Sequencing is performed on two ABI 3700 automated sequencers at the Duke Biology Sequencing Facility. The resulting electrophoreograms are assembled and evaluated using phred and phrap (www.phrap.org).
Each base of a sequence read is assigned a quality value based on the characteristics of the read, the chemistry, and the sequencing hardware. All primer sequences used for sequencing reactions, with information about their respective target genes and their orientation, are stored in the database.
The subsequent contig assembly takes into account the base qualities when assembling the single reads into a contig sequence. Each base of the contig sequence itself is assigned a quality score that can indicate problematic regions and the overall quality of the contig.
Sequences that pass all quality checks (length, quality, blast) are transferred to their respective gene tables.
(3) Sequence verification and local BLAST
Both single sequence reads and assembled contig sequences are subjected to several steps of verification:
- Quality: Each base must remain below a maximum error probability (currently set to 1:1000)
- Length: Contigs must be continuous, and the start and end of new contig’d sequences must also match the reference sequences stored in the database within a given tolerance.
- Origin: Sequences are subjected to a local BLAST search, and their phylum must match the phylum of the first five returned BLAST hits. The BLAST search runs with a custombuilt database that contains all AFToL sequences, all fungal sequences from GenBank, and a random 20% sample of all nonfungal sequences from GenBank.
The sequence authors are informed via email of the results of the BLAST search and the verification, and all related files are available for download through the web interface. If necessary, authors can override any decision made during the automated analysis, and delete sequences or reactivate sequences that did not pass the BLAST analysis.
(4) Alignment (Work in Progress)
Verified gene sequences are automatically aligned to a core alignment. The core alignment contains information about intron positions, ambiguous regions, and other nonalignable elements that are excluded from the alignment process. Starting with the largest block, alignable regions of the core alignment are successively aligned to the new sequence. As a result the sequence is broken down into smaller regions, making the subsequent alignment of the shorter regions easier with each step. This algorithm generates high quality alignments, especially when large amounts of ambiguous regions or introns are present, e.g., in the fungal LSU and SSU nrDNA.
Test for congruence
Prior to the phylogenetic analysis, data sets are tested for congruence. Sequences that cause conflict between data sets are excluded to ensure compatibility between the alignments.
(5) Phylogenetic Analysis
Data sets that have been verified for congruence are analyzed on a regular schedule. Single gene data sets, as well as selected combinations, are analyzed using a variety of methods, such as Maximum Parsimony, Maximum Likelihood, and Bayesian MCMC. Support is estimated with Bootstrap, Bayesian posterior probabilities, and Bayesian Bootstrap. The results are available for the participants of AFToL using the web interface.
Data sets used in publications of AFToL can be downloaded at:
AFToL primers can be found at: